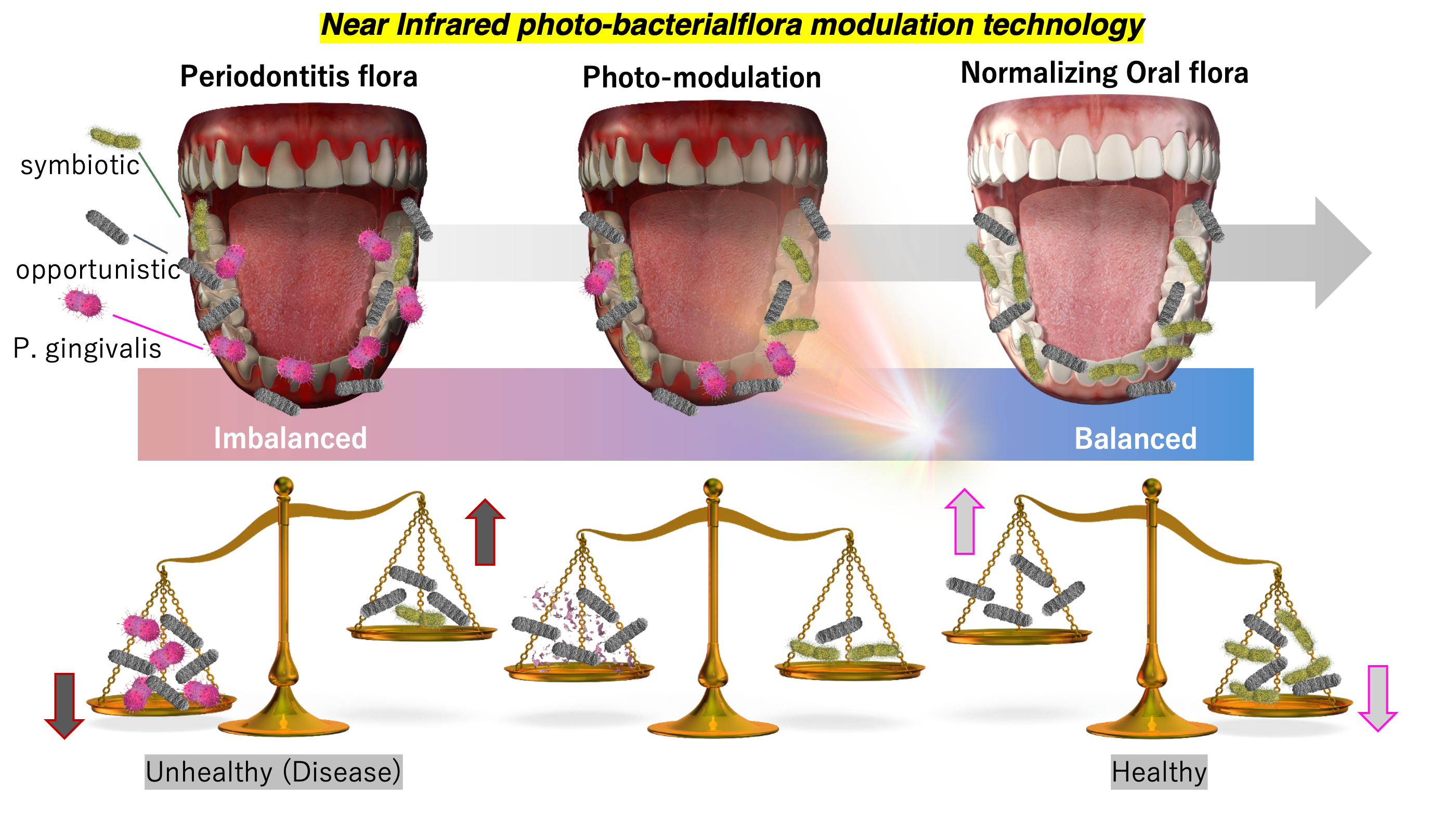
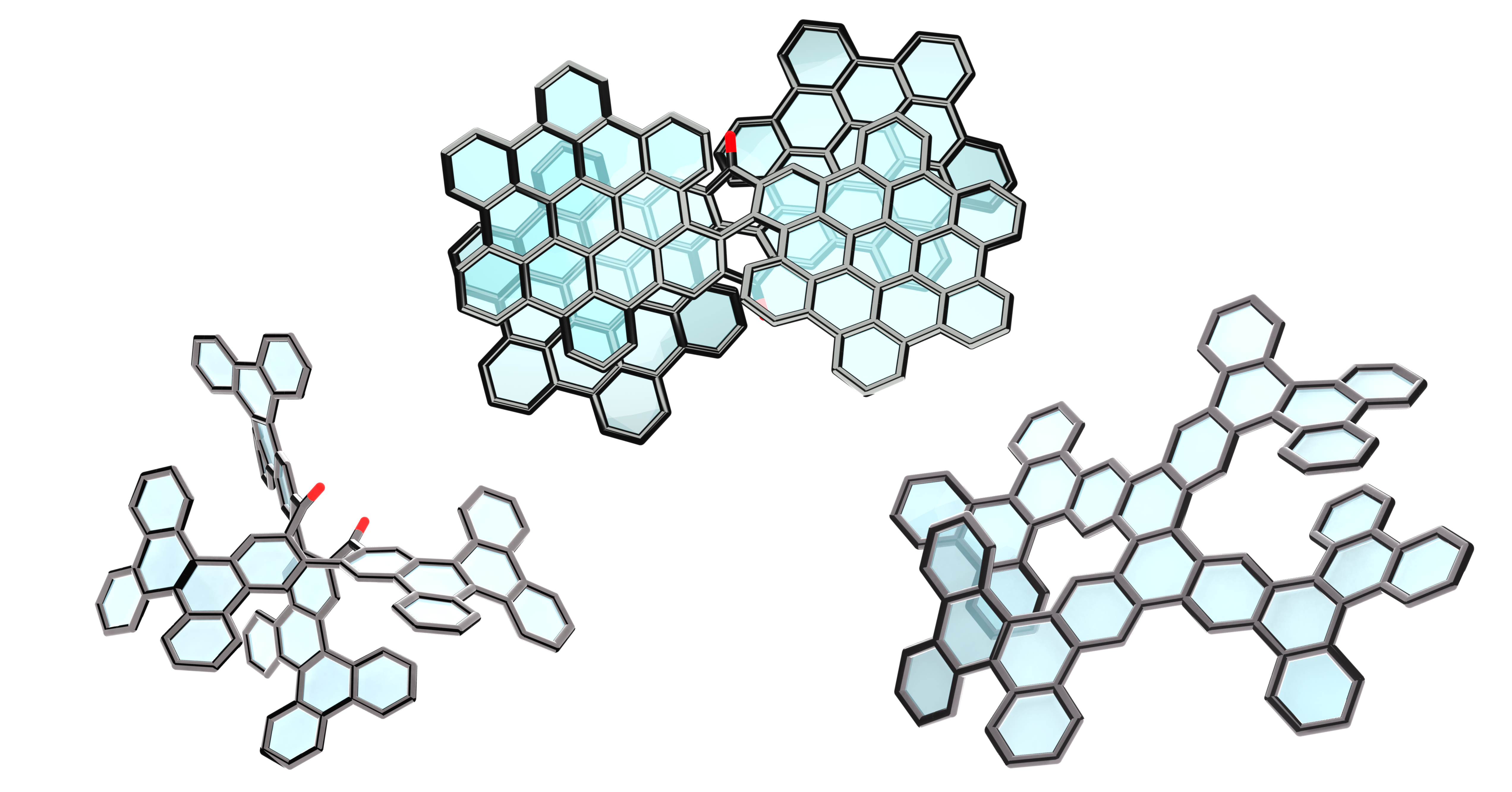
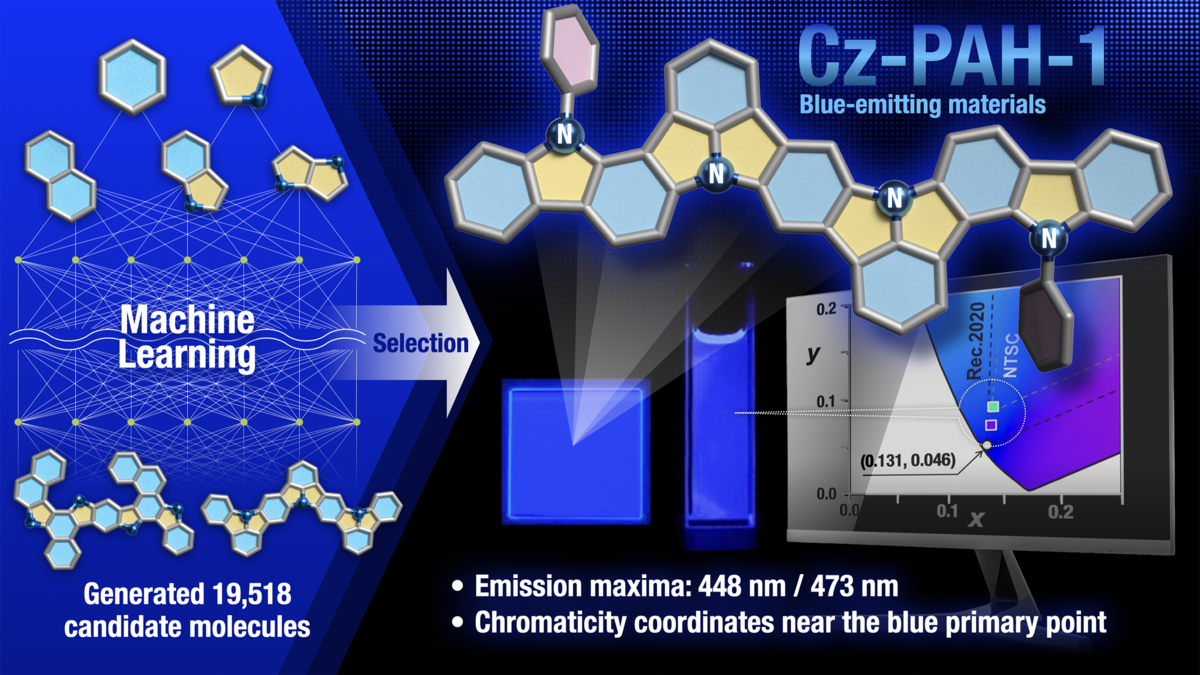
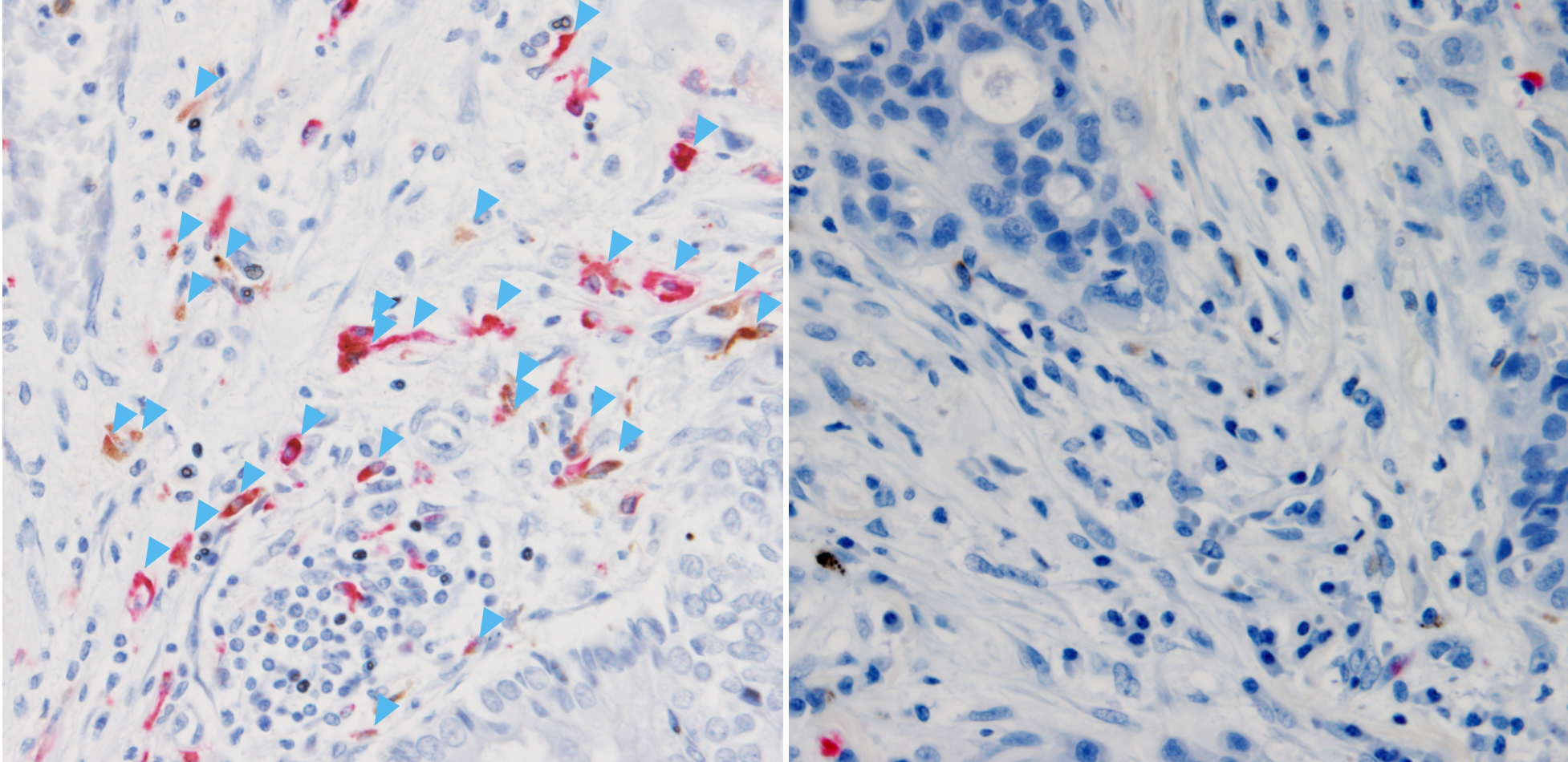
Research & Innovation







We use cookies
By clicking "Accept Cookies," you agree to the use of cookies to improve your user experience, optimize the site, produce statistics, and interact with social networks.
Our Site Policy
News & Events
Stay informed about research breakthroughs, university announcements, and opportunities to engage with Nagoya University's dynamic global community.
Admissions
Study in Japan's fourth largest city, and home to some of its most well-known companies—all without the Tokyo prices and Kyoto crowds.
Academics
Pursue your interests through one of our English or Japanese language programs, selecting from a wide variety of specialized fields.
Research
Learn about our world-class research and comprehensive support systems that empower our researchers to tackle humanity's shared challenges.
Campus life
Find out about our facilities, comprehensive support, extracurricular activities, and the safe and welcoming community that fosters lifelong connections and growth.
About
Meet our leadership and discover the inclusive values and academic heritage that drive Nagoya University's contributions to knowledge and society.
News & Events
Admissions
Academics
Research
Campus life
We use cookies
By clicking "Accept Cookies," you agree to the use of cookies to improve your user experience, optimize the site, produce statistics, and interact with social networks.
Our Site Policy